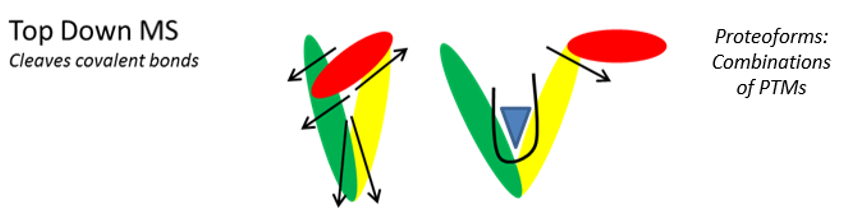
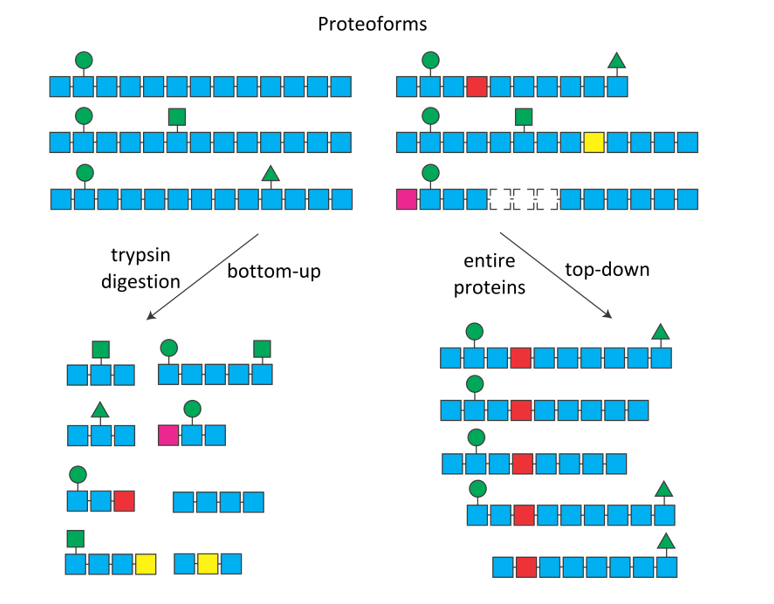
 Figure 1. The combination of post-translational modifications (green symbols), mutations (colored squares) and alternative splicing (dotted squares), can sometimes produce numerous protein isoforms of proteoforms. After digestion (bottom-up, left), it is impossible to know the origin of the trypsic peptides. In top-down (right), each proteoform is selected and fragmented individually.
Figure 1. The combination of post-translational modifications (green symbols), mutations (colored squares) and alternative splicing (dotted squares), can sometimes produce numerous protein isoforms of proteoforms. After digestion (bottom-up, left), it is impossible to know the origin of the trypsic peptides. In top-down (right), each proteoform is selected and fragmented individually.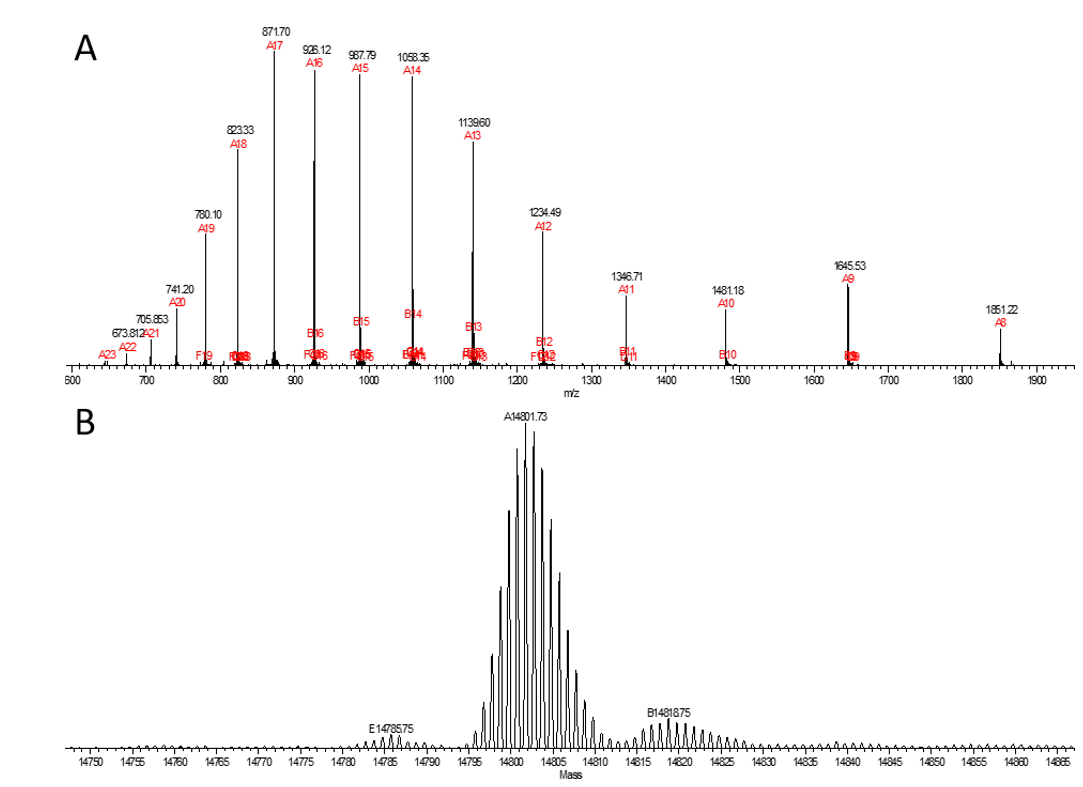
Figure 2. MS spectra corresponding to a ~15 kDa protein.
For more complex mixtures, raw data can be automatically deconvoluted and 3D plots generated, showing the pattern of the most abundant proteins (Figure 3). Targeted top-down analysis can provide information on post-translational modifications (PTMs) and protein identification.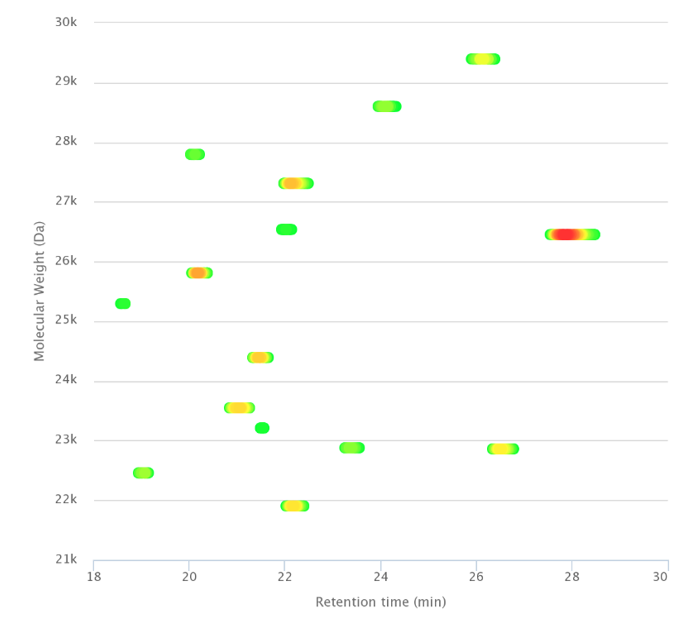
Figure 3. Separation of entire proteins after immunopurification.
2023
Dafun AS, Zivković D, Leon-Icaza SA, Möller S, Froment C, Bonnet D, Almeida de Jesus A, Alric L, Quaranta-Nicaise M, Ferrand A, Cougoule C, Meunier E, Burlet-Schiltz O, Ebstein F, Goldbach-Mansky R, Krüger E, Bousquet Dubouch MP, Marcoux J. Establishing 20S proteasome genetic, translational and post-translational status from precious biological and patient samples with Top-Down MS. Cells. 12, 844.
2022
Živković D, Sanchez Dafun A, Menneteau T, Schahl A, Lise S, Kervarrec C, Toste Rêgo A, da Fonseca PCA, Chavent M, Pineau C, Burlet-Schiltz O, Marcoux J, Bousquet MP. Proteasome complexes experience profound structural and functional rearrangements throughout mammalian spermatogenesis. Proceedings of the National Academy of Sciences. 119(15):e2116826119.
Planès R, Pinilla M, Santoni K, Hessel A, Passemar C, Lay K, Paillette P, Chaves Valadão AL, Samirah Robinson K, Bastard P, Lam N, Fadrique R, Rossi I, Pericat D, Bagayoko S, Adonai Leon-Icaza S, Rombouts Y, Perouzel E, Tiraby M, COVID Human Genetic Effort, Zhang Q, Cicuta P, Jouanguy E, Neyrolles O, Bryant CE, Floto AR, Goujon C, Zhong Lei F, Martin-Blondel G, Silva S, Casanova JL, Cougoule C, Reversade B, Marcoux J, Ravet E, Meunier E. NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Molecular Cell. S1097-2765(22)00433-6.
2020
Lesne J, Locard-Paulet M, Parra J, Zivković D, Menneteau T, Bousquet MP, Burlet-Schiltz O, Marcoux J. Conformational maps of human 20S proteasomes reveal PA28- and immuno-dependant inter-ring crosstalks. Nature Communications 11(1):6140.
2019
Lesne J, Bousquet M-P, Marcoux J and Locard-Paulet M. Top-down and intact protein MS data visualization for proteoform analysis using VisioProt-MS. Bioinformatics and Biology Insights 13:1177932219868223.
2018
Gervais V, Muller I, Mari PO, Mourcet A, Movellan K, Ramos P, Marcoux J, Guillet V, Javaid S, Burlet-Schiltz O, Czaplicki G, Milon A, Giglia-Mari G. Small molecule-based targeting of TTD-A dimerization to control TFIIH transcriptional activity represents a potential strategy for anticancer therapy. Journal of Biological Chemistry 293(39):14974-14988.
Locard-Paulet M, Parra J, Albigot R, Mouton-Barbosa E, Bardi L, Burlet-Schiltz O, Marcoux J. VisioProt-MS: interactive 2D maps from intact protein mass spectrometry. BioInformatics 35(4):679-681.
2017
Parra J, Marcoux J, Poncin I, Canaan S, Herrmann JL, Nigou J, Burlet-Schiltz O, Rivière M. Scrutiny of Mycobacterium tuberculosis 19kDa antigen proteoforms provides new insights in the lipoglycoprotein biogenesis paradigm. Scientific Reports 7:43682.
Carel C, Marcoux J, Parra J, Réat V, Latgé G, Laval F, Burlet-Schiltz O, Demange P, Milon A, Daffé M, Tropis M, Renault M. Post-translational O-mycoloylation mediates protein targeting to the mycomembrane in C. glutamicum. Proceedings of the National Academy of Sciences 114(16) : 4231-4236.