
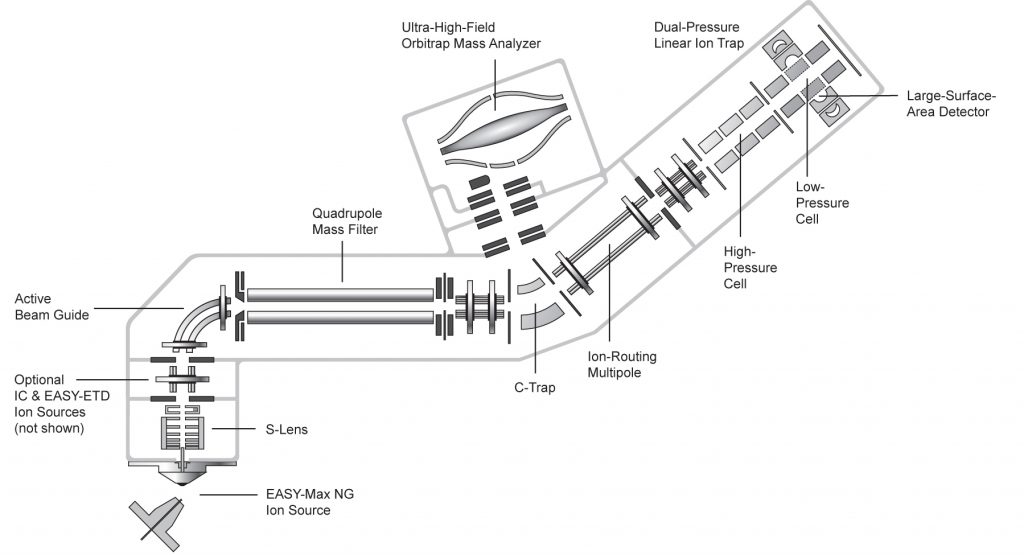
Combining a quadrupole, Orbitrap, and linear ion trap technology in a Tribrid architecture, the Orbitrap Fusion Tribrid mass spectrometer particularly excels at whole-proteome analysis: fulfilling all the requirements of high resolution, mass accuracy and sensitivity necessary for rapid, comprehensive and accurate identification and quantitation of proteins and their post-translational modifications in sample mixtures of low-abundance and high complexity.
- Up to 450,000 resolution (FWHM at m/z 200) enabling molecular-weight determination for intact proteins and confident resolution for isobaric species
- Availability of multiple fragmentation techniques—CID, HCD, and optional ETD and EThCD (EASY-ETD ion source)— at any stage of MSn, with subsequent mass analysis in either the ion trap or Orbitrap mass analyzer for maximum versatility and performance in detailed structural analysis of proteins (e.g. phosphorylation, glycosylation)
- Parallelization of MS and MSn acquisition using a Dynamic Scan Management architecture to maximize the amount of high-quality data acquired
- Synchronous MS3 precursor selection significantly increases the number of peptides accurately quantified in isobaric mass tagging experiments
- Sophisticated data-independent analysis (DIA) and parallel reaction monitoring (PRM) provide reproducible quantitation results while delivering complete qualitative confidence.
Coupled to a UPLC this mass spectrometer is ideal for proteomics applications which include label-free capabilities as well as labelled quantification methods (using SILAC or TMT strategies).
Owing to its ability to acquire spectra at accelerated acquisition speed (high-field Orbitrap mass analyzer) and its extended mass range up to m/z 6000, the Orbitrap Fusion Tribrid mass spectrometer allows to perform top-down experiments with HR/AM MS and MS/MS spectra on intact antibodies and large protein complexes.
| Name | Orbitrap Fusion Tribrid |
|---|---|
|
Company |
Thermo Scientific |
|
Acquisition Date |
2014 |
|
Fragmentation |
CID
HCD
ETD
EThcD (optional EASY-ETD ion source) |
|
Configuration |
Coupled to UHPLC nanoLC systems (Ultimate 3000 RSLCnano, Thermo) |
|
Applications |
Data-Dependent Acquisition
Data-Independent Acquisition
Structural MS Analysis (PTM, Top-down)
Targeted Analysis (PRM) |